Melina2 Help Page
1. What can you do with Melina2?
1) comparison among several motif elucidation programs
Melina2 is a tool which finds potential DNA motifs with a user friendly interface. It runs a maximum of 4 programs from 5 motif prediction tools (Consensus, MEME, Gibbs Sampler, MDScan, Weeder), and returns graphical representations of their results.
2) search against known motifs or genome sequences
Melina2 constructs a weight matrix based on predicted motifs. A user can search possible similar motifs based on these predictions in the database of transcriptional regulation in Bacillus subtilis (DBTBS) or the JASPAR database. Furthermore, with the weight matrix, users can find possible predicted sites in human, mouse, B. subtilis, or E. coli promoter sequences.
3) update in version 2
・ Two prediction programs (MDScan and Weeder) were added, and one (Coreseqnch) was removed because its functionality was essentially the same as Consensus.
・ Users can select 4 statuses in 5 programs with any parameters, and see the results in one glance.
・ The predicted motifs can be illustrated by SequenceLogo.
・ Users can find similar motifs in known transcription factor binding sites motifs in DBTBS or JASPAR.
・ Predicted motif search in several genome sequences is also available.
2.How to use Melina2
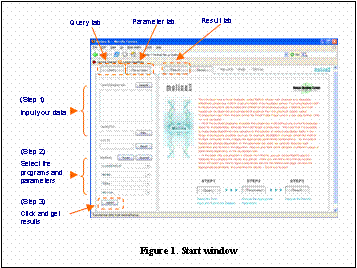 1) welcome screen
1) welcome screen
You can find this screen (Figure 1) when you access Melina2.
You can run Melina2 in 3 simple steps. In fact, the second step can be omitted if you run programs with default parameters.
(Step 1) Input query sequences (FASTA format) (Figure 2)
(Step 2) Set programs and parameters(You can omit this step)
(Step 3) Just run, and get results.
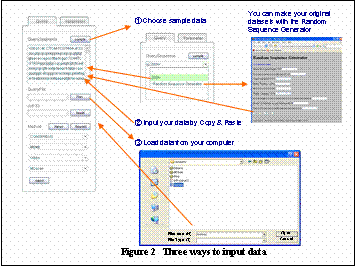 (Step 1) Input query sequences
(Step 1) Input query sequences
There are three ways to load data into Melina2.
1. use sample data
If you click ‘sample’ you can try Melina2 with an example. You can use either 200 bases x 10 sequences or create random sequences using our random sequence generator.
2. use input window
You can copy & paste directly into the input window.
3. load your FASTA formatted file
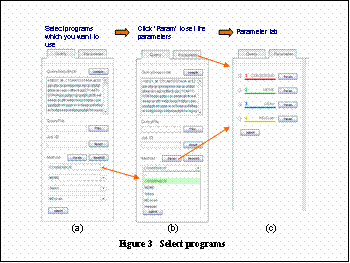 (Step 2) Set programs and parameters
(Step 2) Set programs and parameters
1. choose programs(optional)
You can choose the motif prediction programs. As default Consensus, MEME, Gibbs sampling, and MDscan are selected.
2. set parameters(optional)
Default parameters are not always the same with the original ones supplied by original authors so that the search conditions should be (essentially) the same for all algorithms. We decided a set of new default parameters to make the search condition as similar as possible each other: in principle, (1) the motif length is around 8 bases; (2) both strands are searched; and (3) multiple occurrences are allowed for each sequence. The details are shown in the below table. Fields shown in red mean the places where the values were changed.
| strand | #motifs | size | background | |
| Consensus | both | 0 or more | 10 | NA |
| MEME | both | any | 6-10 | NA |
| Gibbs | both | 0 or more | 10 | NA |
| Weeder | both | some, each | 6,8,10 | H. sapiens |
| MDscan | both | 0 or more | 10 | Query |
To set parameters for each programs, click ‘Param’ in the ‘Query’ tab, or click on the ‘Parameter’ tab(Figure3)
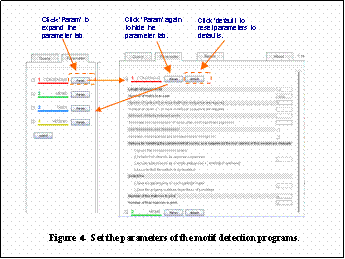 In the ‘Parameter’ tab, click ‘Param’ for each program to set its parameters. (Figure 4)
In the ‘Parameter’ tab, click ‘Param’ for each program to set its parameters. (Figure 4)
Parameters are different for each program, and critical parameters are shown with bold characters. If you click ‘default’, you can revert to our default parameters at any time (they are not always the same with the values set by original authors; see above for more details). Please see the references for more details on each program’s parameters.
(Step 3) Run and get results.
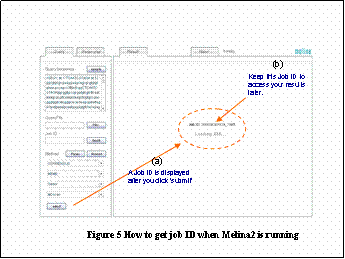 After clicking ‘Submit’, you will see the message “Loading XML” , which means your job is running. If you would like to access your results later, keep the Job ID from the ‘Results’ tab. (Figure5(a))
After clicking ‘Submit’, you will see the message “Loading XML” , which means your job is running. If you would like to access your results later, keep the Job ID from the ‘Results’ tab. (Figure5(a))
After Melina2 finishes motif detection, the results are displayed as shown in Figure 6. Positions of detected motifs are illustrated with colored arrows in the upper window. If you click a motif in the upper window, you can obtain more details in the lower window.
If you would like to know the details for each programs results,
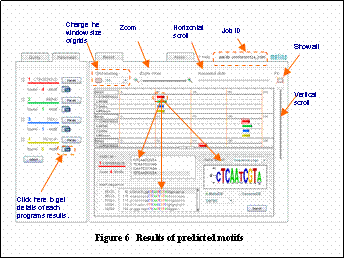 click the ‘Raw’ button next to each program.
click the ‘Raw’ button next to each program.
1) result screen
Detected motifs can be useful to search the motifs in a database of known binding sites or in genome sequence.
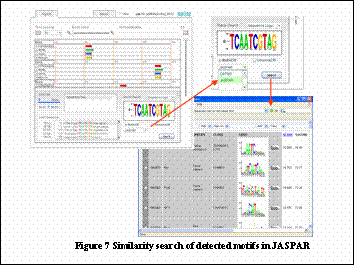
1. Finding similar motifs in known transcription factor binding sites.
Weight matrices of detected motifs are automatically constructed to perform similarity search in JASPAR or DBTBS. The results are shown with SequenceLogo (Figure 7).
2. Putative binding sites search in genome sequences.
User can search putative motifs in human, mouse, B. subtilis, or E. coli genome sequences (Figure 8).

3.Introduction of each program and its main parameters
1) Consensus
Consensus was developed by Stormo et al.[2]. It searches motifs with weight matrix based on information contents.
2) MEME
MEME (Multiple Expectation maximization for Motif Elicitation), which was developed by Bailey et al. [3], predicts no-gap motifs in more than 1 sequences by Expectation maximization algorithm. Even though the original MEME was developed for both amino acids and nucleic acid sequences, Melina2 can accept only DNA sequences. One of three motif models can be selected as a parameter.
OOPS (one occurrence per sequence) model : 1 sequence has 1 motif (default).
ZOOPS (zero or one occurrence per sequence) model : 1 sequence has 0 or 1 motif.
TCM (two-component mixture) model : 1 sequence has 1 or more than 1 motifs.
3) Gibbs Sampling
Gibbs Sampling, which was developed by Lawrence et al. [4], finds statistically significant motifs in given sequences. For example, it first removes one sequence from N given DNA sequences, and randomly selects a motif randomly from each remaining sequences. All random motifs are aligned to construct a weight matrix. At the same time, background nucleic acids frequencies are calculated. Gibbs Sampling then calculates the probability of the motifs with the background model in the previously omitted sequence. It searches again in the remaining sequences based on this probability, and set the position of the motifs.
Then, another sequence is removed and the same steps repeated. Initially, the motifs should be random and meaningless; however, the possibility to detect likely motif is increased if a real motif is chosen by chance. These motifs usually converge to few motifs step by step.
4) MDScan
MDscan, developed by Liu et al [5], searches putative motifs with weight matrices.
5) Weeder
Weeder, invented by Pavesi et al [6], automatically detects motifs in several DNA sequences.
4. About the 'GenomeDB' function
Using a detected motif from Melina II, users can search for occurrences of similar patterns in the upstream (e.g., promoter) regions of all genes in one of 6 species (see below). The search is performed using the 'hmmsearch' program from the HMMER package developed by Sean Eddy (http://hmmer.janelia.org/).
The parameters used are E-value: 10^-5 and SequenceScore: 0.5.
Currently, the following sequences are available.
| #sequences | based on | upstream | downstream | total | source | |
| human | 30964 | TSS | 1000 | 200 | 1200 | DBTSS |
| mouse | 19021 | TSS | 1000 | 200 | 1200 | DBTSS |
| A. thaliana | 30480 | 1st ATG | 1000 | 0 | 1000 | NCBI |
| S. cerevisiae | 5850 | 1st ATG | 800 | 0 | 800 | NCBI |
| E. coli | 4289 | 1st ATG | 300 | 0 | 300 | NCBI |
| B. subtilis | 4100 | 1st ATG | 300 | 0 | 300 | NCBI |
*** Human & mouse
The sequences come from DBTSS (http://dbtss.hgc.jp). The sequences range from 1000 bp upstream to 200 bp downstream of representative transcriptional start sites (TSS) based on DBTSS version 5.2 information.
Note: all alternative promoters in DBTSS are also included.
*** Arabidopsis thaliana
The sequences are constructed based on the entries NC_003070.5, NC_003071.3, NC_003074.4, NC_003075.3, and NC_003076.4 from NCBI. They consist of the 1000 bp upstream of translational start sites (1st ATG).
*** Saccharomyces cerevisiae
The sequences are constructed based on the entries NC_001133.6, NC_001134.7, NC_001135.4, NC_001136.8, NC_001137.2, NC_001138.4, NC_001139.7, NC_001140.5, NC_001141.1, NC_001142.6, NC_001143.7, NC_001144.4, NC_001145.2, NC_001146.5, NC_001147.5 and NC_001148.3. They consist of the 800 bp upstream of translational start sites (1st ATG).
*** Escherichia coli & Bacillus subtilis
The sequences are constructed based on the entries NC_000913.2 and NC_000964.2 from NCBI. They consist of the 300 bp upstream of translational start sites (1st ATG).
5. Caution
・ There is no limitation for input file size; however, the performance depends on the user’s computer environment.
・ We will not take any responsibility for the use of Melina and Melina2, including but not restricted to hardware problems or data loss.
6.References
[1] Poluliakh, N., Takagi and T., Nakai, K. (2003) MELINA : motif extraction from promoter regions of potentially co-regulated genes. Bioinformatics 19(3), pp.423-424
[2] Stormo, G.D. and Hartzell, G.W. (1989) Identifying protein-binding sites from unaligned DNA fragments. Proc. Natl Acad. Sci. USA, 86, pp.1183-1187
[3] Bailey, T.L. and Elkan, C. (1994) Fitting a mixture model by expection maximization to discover motifs
in biopolymers. In Proceedings of 2nd International Conference on Intelligent Systems Molecular Biology.
pp.28-36.
[4]
Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science, 262, pp.208-214
[5] Liu, X.S., Brutlag, D.L. and Liu, J.S. (2002) An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation. Nature Biotech, pp.835-839
[6] Pavesi, G., Mereghetti, P., Mauri, G. and Pesole, G. (2004) WeederWeb: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Research, 32 W199-W203